








一、基态性质计算
首先进行结构优化,结构优化好之后进行扩胞,把CMB和VBM折到Gamma点,因此需要3*3的扩胞,扩胞后的结构和能带如下:

二、NVT计算
优化好结构之后,我们进行非平衡态分子动力学模拟(NVT),使用速度缩放方法(SMASS=-1)使得系统的温度达到300k,用vasp_gam版本进行计算,k点取1*1*1,INCAR设置如下:
#Basic PREC = Normal # PREC= Low | Medium | High | Normal | Accurate | Single ISTART = 0 # Determines whether to read WAVECAR or not. ICHARG = 2 # Determines the 'initial' charge density. LREAL = A # .TRUE.:projection in real space, .FALSE.:reciprocal space. LWAVE = .FALSE. # Written WAVECAR. LCHARG = .FALSE. # Written CHG and CHGCAR. LORBIT = 11 # Written PROCAR and PROOUT. NPAR = 8 # number of cores per compute node. set:sqrt(number of cores) ISYM = 0 # Default # ISPIN = 2 # Default = 1, 2:spin polarized calculations (collinear) are performed. #Electronic Relaxation ENCUT = 400 # Cut-off energy for plane wave basis set in eV. NELM = 200 # Maximum number of electronic SC steps. NELMIN = 6 # Minimum number of electronic SC steps. EDIFF = 1E-5 # Break condition for the electronic SC-loop. GGA = PE # GGA = 91 -- PE -- RP -- PS -- AM #VOSKOWN = 1 # For PW91 and is not required for thr PBE or PBEsol #Ionic Relaxation #NSW = 2000 # Maximum number of ionic steps. # EDIFFG = -0.0002 # Break condition for the ionic relaxation loop IBRION = 0 # Determines how the ions are updated and moved. ISIF = 2 # Controls whether the stress tensor is calculated. POTIM = 1.0 # Scaling constant for the forces. # IVDW = 12 # Add vdW correction to potential energy. #DOS related values ISMEAR = 0 # Determines partial occupancies fnk for each orbital. SIGMA = 0.05 #NBANDS = 112 # Determines the actual number of bands in the calculation. #Molecular dynamics NSW = 4000 # Maximum number of ionic steps. NWRITE = 1 # Determines how much will be written to the file OUTCAR TEBEG = 300 # Start temperature TEEND = 300 # Final temperature SMASS = -1 # -1,0 for NVT and -3 for NVE NBLOCK = 4 # Written to the XDATCAR. ALGO = Normal # Electronic minimisation algorithm MAXMIX = 40 # Maximum number steps stored in Broyden mixer
这一步需要让结构充分弛豫,最终的温度应该在设置的温度附近震荡,可以用grep E= OSZICAR > energy.dat得到温度随时间的数据,用origin画图检查一下:
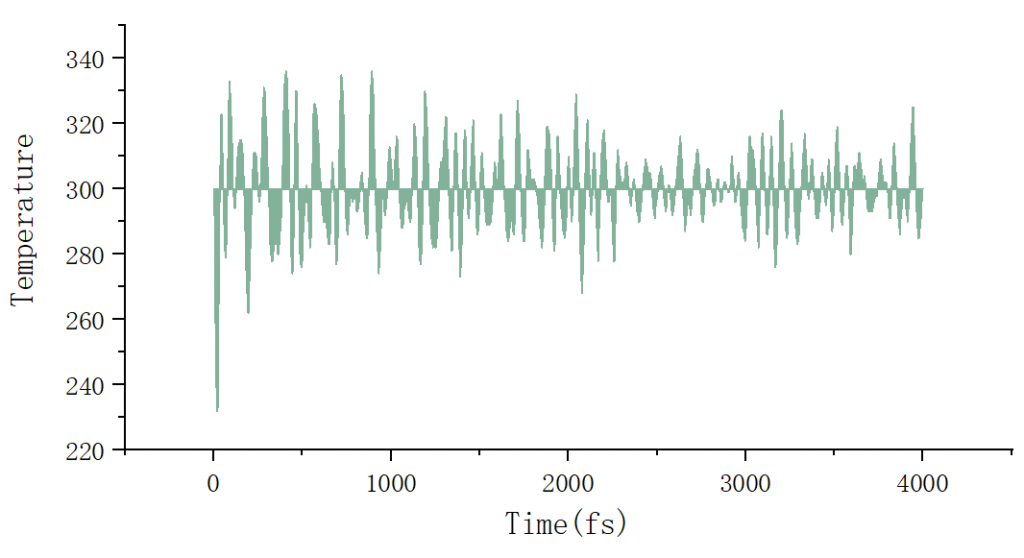
如果温度没有趋于稳定,需要增加md的步数。
三、NVE计算
这一步需要得到分子动力学的轨迹,我们使用微正则系综来进行分子动力学模拟,将上一步的CONTCAR复制为新的POSCAR,并删除速度信息,将SMASS改为-3,即可提交任务,我的INCAR设置如下:
#Basic PREC = Normal # PREC= Low | Medium | High | Normal | Accurate | Single ISTART = 0 # Determines whether to read WAVECAR or not. ICHARG = 2 # Determines the 'initial' charge density. LREAL = A # .TRUE.:projection in real space, .FALSE.:reciprocal space. LWAVE = .FALSE. # Written WAVECAR. LCHARG = .FALSE. # Written CHG and CHGCAR. LORBIT = 11 # Written PROCAR and PROOUT. NPAR = 8 # number of cores per compute node. set:sqrt(number of cores) ISYM = 0 # Default # ISPIN = 2 # Default = 1, 2:spin polarized calculations (collinear) are performed. #Electronic Relaxation ENCUT = 400 # Cut-off energy for plane wave basis set in eV. NELM = 200 # Maximum number of electronic SC steps. NELMIN = 6 # Minimum number of electronic SC steps. EDIFF = 1E-5 # Break condition for the electronic SC-loop. GGA = PE # GGA = 91 -- PE -- RP -- PS -- AM #VOSKOWN = 1 # For PW91 and is not required for thr PBE or PBEsol #Ionic Relaxation #NSW = 2000 # Maximum number of ionic steps. # EDIFFG = -0.02 # Break condition for the ionic relaxation loop IBRION = 0 # Determines how the ions are updated and moved. #ISIF = 2 # Controls whether the stress tensor is calculated. POTIM = 1.0 # Scaling constant for the forces. # IVDW = 12 # Add vdW correction to potential energy. #DOS related values ISMEAR = 0 # Determines partial occupancies fnk for each orbital. SIGMA = 0.05 #NBANDS = 112 # Determines the actual number of bands in the calculation. #Molecular dynamics NSW = 5000 # Maximum number of ionic steps. NWRITE = 1 # Determines how much will be written to the file OUTCAR TEBEG = 300 # Start temperature TEEND = 300 # Final temperature SMASS = -3 # -1,0 for NVT and -3 for NVE NBLOCK = 1 # Written to the XDATCAR. ALGO = Normal # Electronic minimisation algorithm MAXMIX = 40 # Maximum number steps stored in Broyden mixer #Magnetism #NUPDOWN = 2 # Number of electrons #SAXIS = # s_x s_y s_z (quantisation axis for spin) #MAGMOM = # local magnetic moment in x,y,z #Soc related values #LSORBIT = .TRUE. # Switches on spin-orbit coupling. #LNONCOLLINEAR = .TRUE. # Perform fully non-collinear magnetic structure calculations. #LMAXMIX = 4 # Controls up to which l quantum number the onsite PAW charge densities. #GGA_COMPAT = .FALSE. # Restores the full lattice symmetry for gradient corrected functionals. #HSE06 related values #LHFCALC = .TRUE. # Whether Hartree-Fock type calculations are performed. #HFSCREEN = 0.2 # Determines the range separation parameter in hybrid functionals. #ALGO = Damped # Specify the electronic minimisation algorithm. #TIME = 0.4 # Controls the trial time step or the initial (steepest descent) phase. #PRECFOCK = Normal # Controls the FFT grid for the exact exchange (Hartree-Fock) routines. #AEXX = 0.25 # Fraction of exact exchange. #L(S)DA + U #LDAU = .TRUE. #LDAUTYPE = 2 #LDAUL = #LDAUU = #LDAUJ = #LDAUPRINT = 2 #LMAXMIX = 6
四、提取轨迹进行自洽计算
上一步计算完成后,会得到分子动力学的轨迹XDATCAR,这一步我们提取XDATCAR中的结构进行自洽计算,来模拟原子的含时演化。
将XDATCAR复制到一个新的文件夹,并准备xdat2pos.py,直接输入python xdat2pos.py,就会生成一个run文件夹,下面使用一个小脚本进行提交任务,首先准备INCAR,POTCAR,KPOINTS,run.sh,由于只需要计算gamma点,因此KPOINTS只需要设置单k点,自洽计算也只需要用vasp_gam版本,我的run.sh如下:
#!/bin/bash
#SBATCH -J 1
#SBATCH -p bcores48
#SBATCH -n 48
#SBATCH --reservation=qc119_30
module load vasp/5.4.4
module load intel/2017u5
export I_MPI_ADJUST_REDUCE=3
for i in {0001..0570};do
cd run/$i
ln -sf ../../INCAR
ln -sf ../../KPOINTS
ln -sf ../../POTCAR
#ln -sf ../../vasp.sh
mpirun -np 48 vasp_gam &>log
cd ../..
done
五、计算NAC
在run文件夹中,准备input.py,以及Dephase.py,其中input.py如下:
#!/usr/bin/env python
# -*- coding: utf-8 -*-
from CAnac import nac_calc
T_start = 1
T_end = 4000
# NAC calculations and Genration of standard input for HFNAMD or PYXAID
# bmin and bmax are actual band index in VASP,
# and should be same with the bmin and bmax in your NAMD simulation.
is_combine = True #If generate standard input for HFNAMD or PYXAID
#iformat = "PYXAID"
iformat = "HFNAMD"
bmin = 81
bmax = 82
potim = 1 # Nuclear timestep, unit: fs
# Time-overlap
# bmin_stored bmax_stored are actual band index in VASP
# Use a large basis sets here if
# you would like to remove WAVECAR to save disk usage
# Or when you turn on the state reordering
# bmin_stored = bmin - 10
# bmax_stored = bmax + 10
bmin_stored = 75
bmax_stored = 85
nproc = 4 # Number of cores used in parallelization
is_gamma_version = True # Which VASP version is used!!
# vasp_std False vasp_gam True
is_reorder= False # If turn on State Reordering
# True (use with care) or False
is_alle = False # If use All-electron wavefunction
# (require NORMALCAR) True or False
is_real = True # If rotate wavefunction to ensure NAC is real value.
# True (Mandatory for HFNAMD and PYXAID) or False.
ikpt = 1 #k-point index, starting from 1 to NKPTS
ispin = 1 #spin index, 1 or 2
# Directories structure.
# Here, 0001 for 1st ionic step, 0002 for 2nd ionic step, etc.
# Don't forget the forward slash at the end.
Dirs = ['./%04d/' % (ii + 1) for ii in range(T_start-1, T_end)]
# Don't change anything below if you are new to CA-NAC
#########################################################################
# For Pseudo NAC only. omin and omax are used for post-orthonormalization.
# In principle, you should use entire basis sets in VASP
icor = 1
omin = bmin_stored
omax = bmax_stored
skip_file_verification = False
skip_TDolap_calc = False
skip_NAC_calc = False
onthefly_verification = True
checking_dict={'skip_file_verification':skip_file_verification,
'skip_TDolap_calc':skip_TDolap_calc,
'skip_NAC_calc':skip_NAC_calc,
'onthefly_verification':onthefly_verification}
nac_calc(Dirs, checking_dict, nproc,
is_gamma_version, is_reorder, is_alle, is_real, is_combine,
iformat, bmin, bmax,
bmin_stored, bmax_stored, omin, omax,
ikpt, ispin, icor, potim )
其中T_end是上一步进行自洽计算的步数,bmin = 81,bmax = 82 为VBM和CBM的序号,运行之后会产生如下文件:
依次运行以下命令:
cp CAeig_81_82_ispin1_k1_ps_real.txt energy.dat python Dephase.py
这时会产生一个DEPHTIME文件,用以描述退相干作用。
六、NAMD计算
将CAnac_81_82_ispin1_k1_ps_real_re.txt命名为NATXT,CAeig_81_82_ispin1_k1_ps_real.txt改为EIGTXT,连同DEPHTIME一起复制到一个新的文件夹中,准备inp以及INICON,提交hfnamd任务,其中inp设置如下:
&NAMDPARA BMIN = 81 BMAX = 82 NSAMPLE = 30 NTRAJ = 500 NSW = 3998 NELM = 10 TEMP = 300 NAMDTIME = 300000 POTIM = 1.0 ALGO = "DISH" ALGO_INT = 0 LHOLE = .FALSE. LSHP = .TRUE. LCPTXT = .TRUE. DEBUGLEVEL = "I" /
七、拟合函数计算载流子寿命
上一步计算完成之后,会得到一系列SHPROP文件,文件格式如下:

其中最后一列就是电子的占据数,我们对所有的SHPROP求平均,第一列为横坐标画图,用origin拟合函数y=exp(-x/A),A就是拟合得到的载流子寿命,最终效果如下:
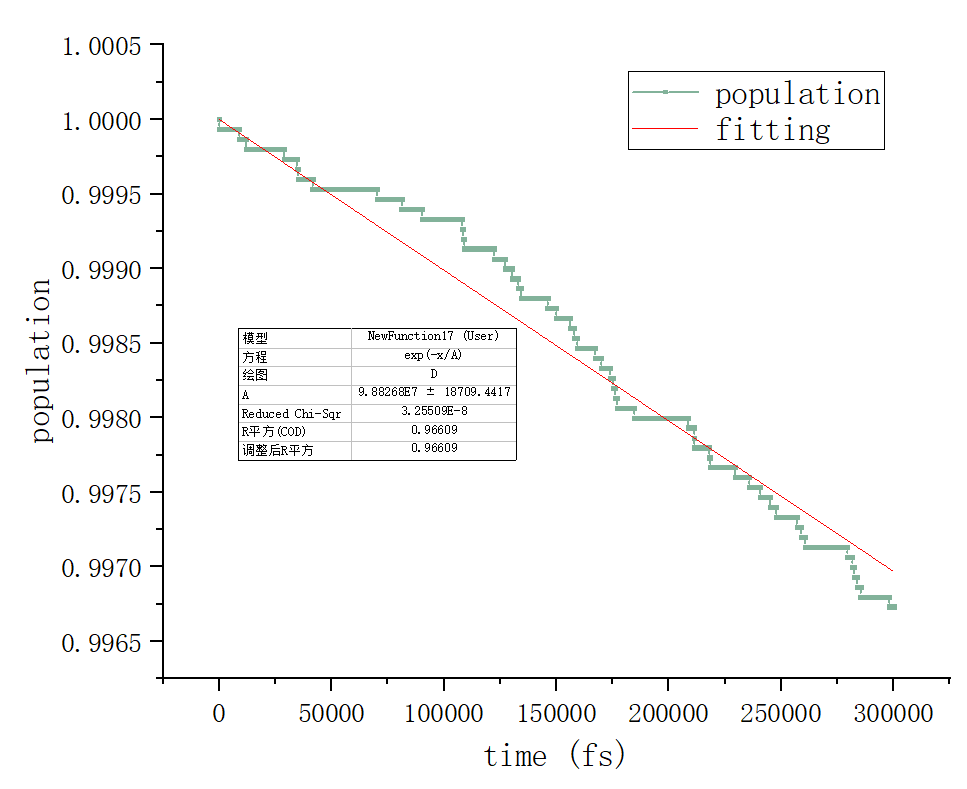
最终计算结果为98ns左右,到这里计算就全部完成了!
No Comments
Leave a comment Cancel