








以石墨烯为例使用cp2k计算能带
第一步创建输入文件,可以用Multiwfn快速创建,可以避免设置很多繁琐的参数,Multiwfn下载网站如下,这里我们使用的是windows版本的。
软件解压后可直接运行,将石墨烯的cif文件放在解压后的文件夹内,cif文件可以自己网上找,有很多。双击打开Multiwfn.exe,运行后如下图所示:
依次输入以下命令:
./Graphene.cif cp2k ENTER 8 #设置自洽k点密度 11,11,1 0 q #推出程序
运行结束后会在当前文件夹下出现Graphene.inp文件,这就是cp2k的输入文件,接下来编辑这个文件,手动设置k点路径以及沿着k点路径撒点密度,如下图所示:
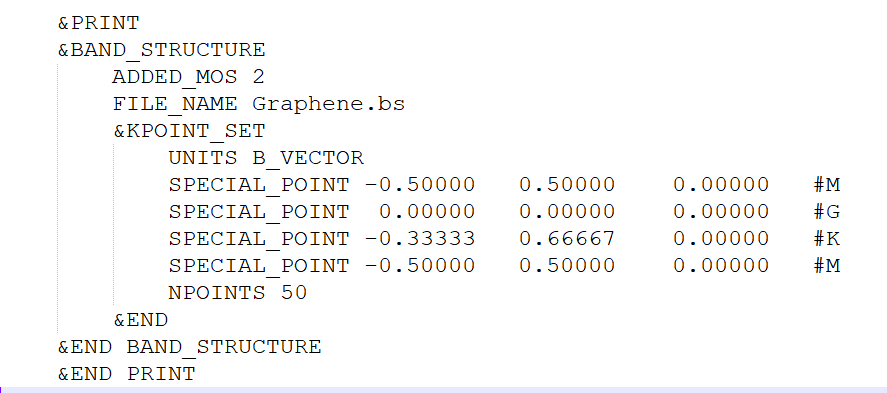
此外,需要注意的是直接计算并不会输出费米能级,需要设置如下参数:
&SMEAR ON
METHOD FERMI_DIRAC # 或者其他的smearing方法,如GAUSSIAN
ELECTRONIC_TEMPERATURE [K] 0 # 以Kelvin为单位,设置电子温度
&END SMEAR
ADDED_MOS 20
完整的输入文件如下:
#Generated by Multiwfn
&GLOBAL
PROJECT Graphene
PRINT_LEVEL LOW
RUN_TYPE ENERGY
&END GLOBAL
&FORCE_EVAL
METHOD Quickstep
&SUBSYS
&CELL
A 2.46515800 0.00000000 0.00000000
B -1.23257017 2.13489455 0.00000000
C 0.00000000 0.00000000 15.00000000
PERIODIC XYZ #Direction(s) of applied PBC (geometry aspect)
&END CELL
&COORD
C 1.23196688 0.71056336 7.50000000
C -0.00061164 1.42219630 7.50000000
&END COORD
&KIND C
ELEMENT C
BASIS_SET DZVP-MOLOPT-SR-GTH-q4
POTENTIAL GTH-PBE
&END KIND
&END SUBSYS
&DFT
BASIS_SET_FILE_NAME BASIS_MOLOPT
POTENTIAL_FILE_NAME POTENTIAL
# WFN_RESTART_FILE_NAME Graphene-RESTART.kp
CHARGE 0 #Net charge
MULTIPLICITY 1 #Spin multiplicity
&KPOINTS
SCHEME MONKHORST-PACK 11 11 1
&END KPOINTS
&PRINT
&BAND_STRUCTURE
ADDED_MOS 2
FILE_NAME Graphene.bs
&KPOINT_SET
UNITS B_VECTOR
SPECIAL_POINT -0.50000 0.50000 0.00000 #M
SPECIAL_POINT 0.00000 0.00000 0.00000 #G
SPECIAL_POINT -0.33333 0.66667 0.00000 #K
SPECIAL_POINT -0.50000 0.50000 0.00000 #M
NPOINTS 50
&END
&END BAND_STRUCTURE
&END PRINT
&QS
EPS_DEFAULT 1.0E-11 #Set all EPS_xxx to values such that the energy will be correct up to this value
&END QS
&POISSON
PERIODIC XYZ #Direction(s) of PBC for calculating electrostatics
PSOLVER PERIODIC #The way to solve Poisson equation
&END POISSON
&XC
&XC_FUNCTIONAL PBE
&END XC_FUNCTIONAL
&END XC
&MGRID
CUTOFF 350
REL_CUTOFF 50
&END MGRID
&SCF
MAX_SCF 128
EPS_SCF 5.0E-06 #Convergence threshold of density matrix of inner SCF
# SCF_GUESS RESTART #Use wavefunction from WFN_RESTART_FILE_NAME file as initial guess
# IGNORE_CONVERGENCE_FAILURE #Continue calculation even if SCF not converged, works for version >= 2024.1
&DIAGONALIZATION
ALGORITHM STANDARD #Algorithm for diagonalization
&END DIAGONALIZATION
&MIXING #How to mix old and new density matrices
METHOD BROYDEN_MIXING #PULAY_MIXING is also a good alternative
ALPHA 0.4 #Default. Mixing 40% of new density matrix with the old one
NBROYDEN 8 #Default is 4. Number of previous steps stored for the actual mixing scheme
&END MIXING
&SMEAR ON
METHOD FERMI_DIRAC # 或者其他的smearing方法,如GAUSSIAN
ELECTRONIC_TEMPERATURE [K] 0 # 以Kelvin为单位,设置电子温度
&END SMEAR
ADDED_MOS 20
&END SCF
&END DFT
&END FORCE_EVAL
可以看到,相比于Multiwfn自动生成的,仅仅修改了k点路径以及费米能级相关参数,接下来就可以提交任务计算了。
cp2k的计算速度非常快,几秒钟应该就能算完,算完之后会得到Graphene.bs文件,这就是我们需要的能带数据。
同时,我们可以查看输出文件,搜索Fermi可以找到费米能级,需要注意的是这里面的能量是原子单位制,需要化为eV,直接将数值乘以27.211386245988即可。
最后就是用这个数据画出能带图,由于没有什么方便的软件,这里用python写了一个小脚本,在windows上直接运行即可,脚本如下:
import matplotlib.pyplot as plt
import re
# 读取文件内容
file_path = './Graphene.bs' # 请替换为您的文件路径
with open(file_path, 'r') as file:
content = file.readlines()
# 初始化数据存储结构
data_refined = {}
current_key_refined = None
Fermi = -27.211386245988*0.07967043079508
# 处理文件内容,提取数据
for line in content:
if "Nr." in line:
# 使用正则表达式精确提取Nr.后的数字作为键
current_key_refined = float(re.search(r'Nr\.\s+(\d+)', line).group(1))
data_refined[current_key_refined] = []
elif current_key_refined is not None:
try:
# 尝试将行内的每个分段转换为浮点数,并将其添加到当前键的列表中
y_values = [float(y) - Fermi for y in line.split()]
if y_values: # 确保列表不为空
data_refined[current_key_refined].extend(y_values)
except ValueError:
# 如果转换失败,则跳过此行
continue
# 绘制图表
plt.figure(figsize=(10, 6))
for x, y_values in data_refined.items():
plt.scatter([x] * len(y_values), y_values,color='b', label=f'Nr. {x}', s=10)
plt.xlabel('Band Index')
plt.ylabel('Energy (eV)')
plt.ylim(-5,5)
plt.title('Band Structure')
#plt.legend(markerscale=2)
plt.grid(True)
# 添加垂直虚线,统一使用蓝色
plt.axvline(x=51, color='b', linestyle='--') # 移除label以避免重复
plt.axvline(x=101, color='b', linestyle='--')
plt.show()
print(data_refined.items())
# 输出数据到文件
with open('band.dat', 'w') as f:
for x, y in data_refined.items():
f.write(f'{x}'+' ')
for y in y_values:
f.write(f'{y}'+' ')
f.write('\n')
运行之后得到如下图片:
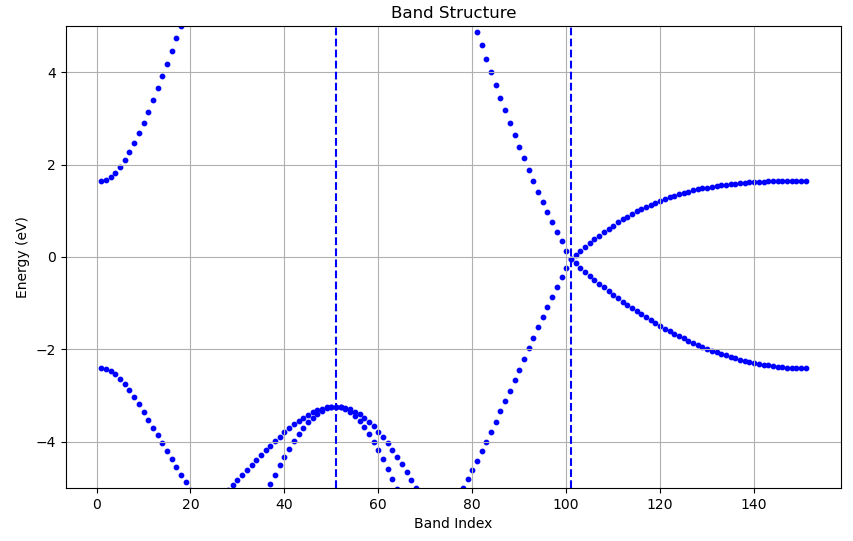
由于时间原因,这个脚本写的比较粗糙,嫌丑的自行修改,到这里能带就计算完成了,可以看到和vasp结果一致。
No Comments
Leave a comment Cancel