








这是一个单分子的非绝热动力学的例子,可以用来快速掌握计算方法,这里使用的QE版本为6.5
第一步:使用QE进行分子动力学模拟
准备QE的输入文件以及赝势,输入文件如下:
&CONTROL
calculation = 'md',
dt = 20.67055,
nstep = 5000,
pseudo_dir = './',
outdir = './',
prefix = 'x',
disk_io = 'low',
/
&SYSTEM
ibrav = 0,
celldm(1) = 1.89,
nat = 6,
ntyp = 2,
nspin = 1,
nbnd = 20,
ecutwfc = 30,
tot_charge = 0.0,
occupations = 'smearing',
smearing = 'gaussian',
degauss = 0.005,
nosym = .true.,
/
&ELECTRONS
electron_maxstep = 300,
conv_thr = 1.D-5,
mixing_beta = 0.45,
/
&IONS
ion_dynamics = 'verlet',
ion_temperature = 'andersen',
tempw = 300.00 ,
nraise = 1,
/
ATOMIC_SPECIES
C 12.01 C.pbe-rrkjus.UPF
H 1.008 H.pbe-rrkjus.UPF
K_POINTS gamma
CELL_PARAMETERS
10.0000000 0.0000000 0.0000000
0.0000000 10.0000000 0.0000000
0.0000000 0.0000000 10.0000000
ATOMIC_POSITIONS (alat)
C -5.04672 2.31763 0.01782
C -3.71548 2.30894 -0.00587
H -5.60642 1.54073 -0.49255
H -5.57772 3.10164 0.54761
H -3.18448 1.52493 -0.53565
H -3.15579 3.08584 0.50451
参数详细说明: &CONTROL: calculation = 'md': 指定计算类型;在此情况下,是分子动力学(MD)模拟。 dt = 20.67055: MD模拟的时间步长,单位通常是原子单位(au),1 a.u. = 4.8378 * 10 ^ -17 s,相当于1fs。 nstep = 5000: MD步数。 pseudo_dir = './': 赝势文件所在的目录。 outdir = './': 输出文件的目录。 prefix = 'x': 输出文件名的前缀。 disk_io = 'low': 指定I/O活动的级别。 &SYSTEM : ibrav = 0: 布拉维格子类型(0表示晶胞由celldm定义)。 celldm(1) = 1.89: 第一个celldm参数,指定单位晶胞的整体缩放。 nat = 6: 单位晶胞中的原子数。 ntyp = 2: 原子类型的数量。 nspin = 1: 自旋分量的数量。 nbnd = 20: 电子带的数量。 ecutwfc = 30: 波函数的能量截断。 tot_charge = 0.0: 系统的总电荷。 occupations = 'smearing': 指定电子占据的处理方式。 smearing = 'gaussian': 使用的平滑类型。 degauss = 0.005: 平滑的宽度,以电子伏特为单位。 nosym = .true.: 禁用对称性。 &ELECTRONS: electron_maxstep = 300: 电子步数的最大值。 conv_thr = 1.D-5: 电子自洽性的收敛阈值。 mixing_beta = 0.45: 控制电荷密度混合的参数。 &IONS: ion_dynamics = 'verlet': 指定离子动力学的积分算法(Verlet算法)。 ion_temperature = 'andersen': 指定控制离子温度的恒温器方法(Andersen恒温器)。 tempw = 300.00: 恒温器的目标温度。 nraise = 1: 在速度重新缩放尝试之间的步数。
赝势文件到下面的网站下载,注意赝势的文件名称和路径要与输入文件一致:
PSlibrary – QUANTUMESPRESSO (quantum-espresso.org)
最后是QE的提交脚本,可以看到当前文件夹中有以下文件:
> ls ] C.pbe-rrkjus.UPF H.pbe-rrkjus.UPF submit.slm x.md.in
运行过程中,可以通过查看文件x.msd来看运行了多少步:
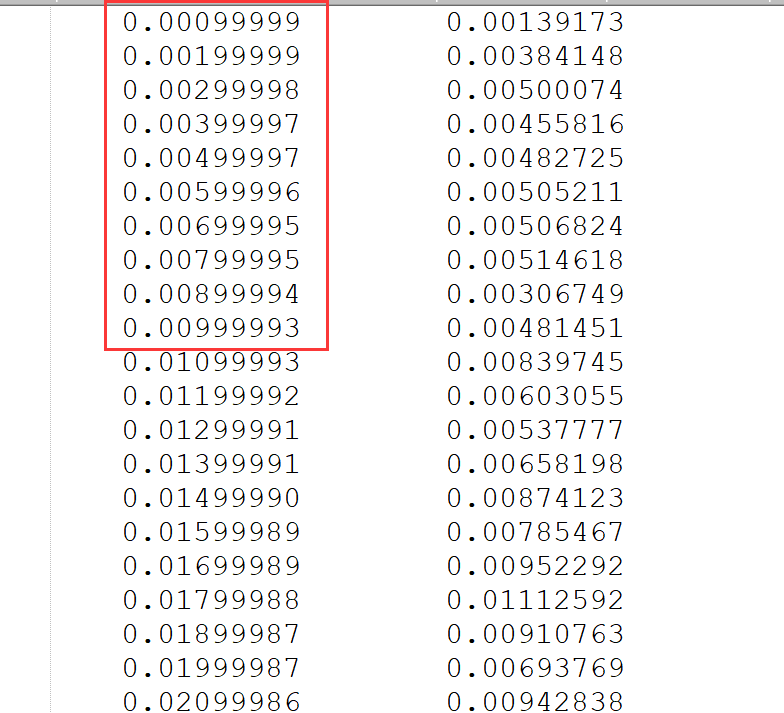
第一列就是运行的时间步,单位是皮秒(ps)
第二步:计算电子的哈密顿量
将上一步分子动力学产生的文件复制到一个新的文件夹step2中
准备赝势文件以及输入文件模板x0.exp.in及x0.scf.in:
&inputpp prefix = 'x0', outdir = './', pseudo_dir = '/data/home/qc119/workspace/xiaqian/test_pyxaid/Tut2_small/step2', psfile(1) = 'C.pbe-rrkjus.UPF', psfile(2) = 'H.pbe-rrkjus.UPF', single_file = .FALSE., ascii = .TRUE., uspp_spsi = .FALSE., /
&CONTROL
calculation = 'scf',
pseudo_dir = '/data/home/qc119/workspace/xiaqian/test_pyxaid/Tut2_small/step2',
outdir = './',
prefix = 'x0',
disk_io = 'low',
wf_collect = .true.
/
&SYSTEM
ibrav = 0,
celldm(1) = 1.89,
nat = 6,
ntyp = 2,
nspin = 2,
nbnd = 20,
ecutwfc = 30,
tot_charge = 0.0,
starting_magnetization(1) = 0.01
occupations = 'smearing',
smearing = 'gaussian',
degauss = 0.005,
nosym = .true.,
/
&ELECTRONS
electron_maxstep = 300,
conv_thr = 1.D-5,
mixing_beta = 0.45,
/
&IONS
ion_dynamics = 'verlet',
ion_temperature = 'andersen',
tempw = 300.00 ,
nraise = 1,
/
ATOMIC_SPECIES
C 12.01 C.pbe-rrkjus.UPF
H 1.008 H.pbe-rrkjus.UPF
K_POINTS gamma
CELL_PARAMETERS
10.0000000 0.0000000 0.0000000
0.0000000 10.0000000 0.0000000
0.0000000 0.0000000 10.0000000
这一步注意设置统一的赝势文件路径,避免多次复制赝势文件
提交脚本模板,最上面需要根据你使用的集群修改:
#!/bin/bash
#SBATCH -J xiaqian
#SBATCH -p bcores48
#SBATCH -n 48
#SBATCH --reservation=qc119_30
module load intel/2017u5
export I_MPI_ADJUST_REDUCE=3
echo "SLURM_JOBID="$SLURM_JOBID
echo "SLURM_JOB_NODELIST="$SLURM_JOB_NODELIST
echo "SLURM_NNODES="$SLURM_NNODES
echo "SLURMTMPDIR="$SLURMTMPDIR
echo "working directory="$SLURM_SUBMIT_DIR
NPROCS=`srun --nodes=${SLURM_NNODES} bash -c 'hostname' |wc -l`
echo NPROCS=$NPROCS
module load gcc/7.5.0
# Setup all manual parameters here
# Must be ABSOLUTE paths
NP=1
exe_qespresso=/data/home/qc119/sourcecode/QE6.5/qe-6.5/bin/pw.x
exe_export=/data/home/qc119/sourcecode/QE6.5/qe-6.5/bin/pw_export.x
exe_convert=/data/home/qc119/sourcecode/QE6.5/qe-6.5/bin/iotk
res=/data/home/qc119/workspace/xiaqian/test_pyxaid/Tut2_small/step2/res
#上面的内容都要根据具体情况修改,下面的不需要修改
# These will be assigned automatically, leave them as they are
param1=
param2=
# This is invocation of the scripts which will further handle NA-MD calclculations
# on the NAC calculation step
python -c "from PYXAID import *
params = { }
params[\"NP\"]=$NP
params[\"EXE\"]=\"$exe_qespresso\"
params[\"EXE_EXPORT\"]=\"$exe_export\"
params[\"EXE_CONVERT\"] = \"$exe_convert\"
params[\"start_indx\"]=$param1
params[\"stop_indx\"]=$param2
params[\"wd\"]=\"wd_test\"
params[\"rd\"]=\"$res\"
params[\"minband\"]=4
params[\"nocc\"]=6
params[\"maxband\"]=10
params[\"nac_method\"]=0
params[\"wfc_preprocess\"]=\"complete\"
params[\"do_complete\"]=1
params[\"prefix0\"]=\"x0.scf\"
params[\"prefix1\"]=\"x1.scf\"
params[\"compute_Hprime\"]=0
print params
runMD1.runMD(params)
"
脚本py-scr2.py:
from PYXAID import *
import os
nsteps_per_job = 50
tot_nsteps = 500
# Step 1 - split MD trajectory on the time steps
# Provide files listed below: "x.md.out" and "x.scf.in"
# IMPORTANT:
# 1) use only ABSOLUTE path for PP in x.scf.in file
# 2) provided input file is just a template, so do not include coordinates
out2inp.out2inp("x.md.out","x0.scf.in","wd","x0.scf",0,tot_nsteps,1) # neutral setup
#out2inp.out2inp("x.md.out","x1.scf.in","wd","x1.scf",0,tot_nsteps,1) # charged setup
# Step 2 - distribute all time steps into groups(jobs)
# several time steps per group - this is to accelerate calculations
# creates a "customized" submit file for each job and submit it - run
# a swarm in independent calculations (trajectory pieces)
# (HTC paradigm)
# Provide the files below:
# submit_templ.pbs - template for submit files - manually edit the variables
# x.exp.in - file for export of the wavefunction
os.system("cp submit_templ.slm wd")
os.system("cp x0.exp.in wd")
#os.system("cp x1.exp.in wd")
os.chdir("wd")
distribute.distribute(0,tot_nsteps,nsteps_per_job,"submit_templ.slm",["x0.exp.in"],["x0.scf"],2)
#distribute.distribute(0,tot_nsteps,nsteps_per_job,"submit_templ.pbs",["x0.exp.in","x1.exp.in"],["x0.scf","x1.scf"],1)
其中, nsteps_per_job = 50 tot_nsteps = 500 代表计算500步,分为10部分计算,每部分计算50步
当前目录下有以下文件:
> ls ] C.pbe-rrkjus.UPF H.pbe-rrkjus.UPF py-scr2.py submit_templ.slm x0.exp.in x0.scf.in x.md.out
创建输出结果的文件夹,并运行脚本,自动提交任务:
> mkdir res > python py-scr2.py
运行过程中,可以使用脚本py-scr7.py来时事监控任务进程,直接输入命令python py-scr7.py即可,也可以看res目录是否产生了结果文件,这一步也是时事更新的,如果没有文件,可能就是出错了,可以运行命令看一下:
> python py-scr7.py ] range(0,178) nelts = 178 > ls ./res ] 0_Ham_0_im 0_Ham_119_re 0_Ham_139_im 0_Ham_158_re 0_Ham_24_im 0_Ham_43_re 0_Ham_63_im 0_Ham_82_re 0_Ham_0_re 0_Ham_11_im 0_Ham_139_re 0_Ham_159_im 0_Ham_24_re 0_Ham_44_im 0_Ham_63_re 0_Ham_83_im 0_Ham_100_im 0_Ham_11_re 0_Ham_13_im 0_Ham_159_re 0_Ham_25_im 0_Ham_44_re 0_Ham_64_im 0_Ham_83_re 0_Ham_100_re 0_Ham_120_im 0_Ham_13_re 0_Ham_15_im 0_Ham_25_re 0_Ham_45_im 0_Ham_64_re 0_Ham_84_im
接下来在step2中创建文件夹spectr,并运行脚本py-scr6.py,可以看到spectr中出现了一些文件:
> mkdir spectr > python py-scr6.py > cd spectr > ls ] 1ave_Ham_im.dat 1ave_Ham_re.dat ave_Ham_im.dat ave_Ham_re.dat
最后,创建一个文件夹,进行namd的计算,命名为step3,在step3文件夹中创建目录:
> mkdir out > mkdir macro
直接运行脚本py-scr3.py:
python py-scr3.py
最后运行脚本py-scr4.py进行数据处理,结果放在文件夹macro中:
> cd macro/ > ls ] energy_macro_ex0 plt_td_map se_en_ex0 se_pop_ex0 se_pop_macro_ex1 sh_en_ex1 sh_pop_ex1 energy_macro_ex1 relax.png se_en_ex1 se_pop_ex1 sh.dat sh_map.png sh_pop_macro_ex0 plt_relax se.dat se_map.png se_pop_macro_ex0 sh_en_ex0 sh_pop_ex0 sh_pop_macro_ex1
将脚本plt_relax和plt_td_map复制到当前文件夹,分别运行这两个脚本:
> gnuplot plt_relax > gnuplot plt_td_map
结果如下:
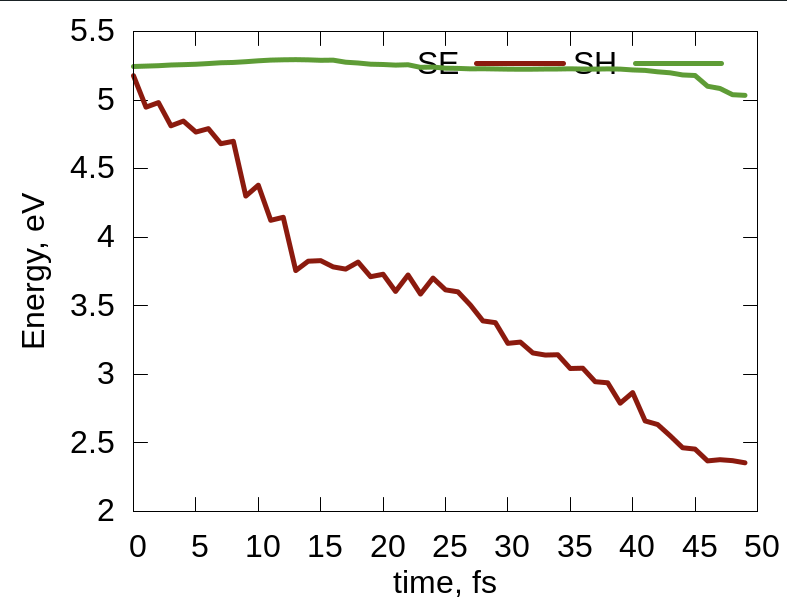
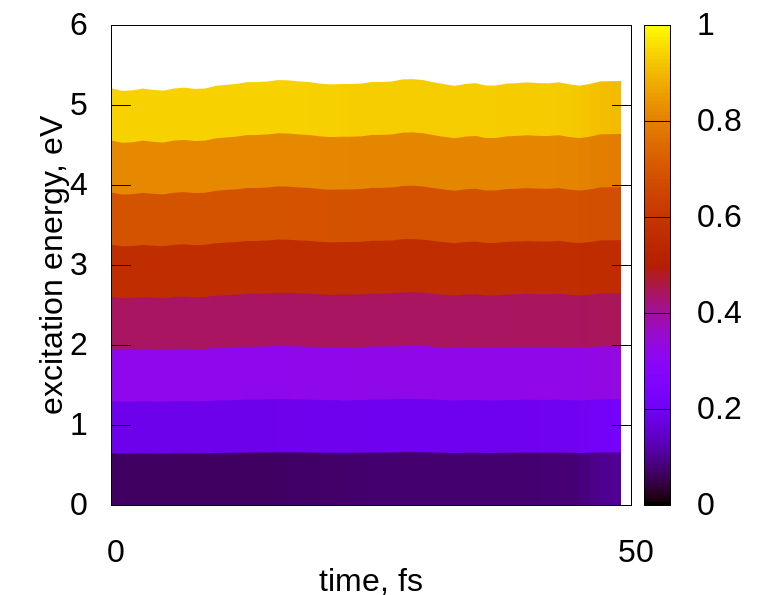
No Comments
Leave a comment Cancel