








一、软件的安装
其实之前写过如何安装hefei-NAMD,但是由于软件的迭代更新,安装步骤也有了一些变化,这里我们重新安装一遍。 首先输入以下命令,获取安装包:
git clone https://github.com/realxiangjiang/paraHFNAMD
运行之后会出现以下文件:
接下来激活intel编译器,需要Intel2021及以上的版本,不然会报错,如果没有安装可以联系你们自己集群的管理员进行安装,安装好之后激活编译器,这里我们集群用以下命令:
module load gcc/7.5.0 source /data/software/intel/2022u2/setvars.sh intel64
激活编译器之后进入目录:paraHFNAMD/src,直接输入make,就可以正常安装。 安装成功之后,在paraHFNAMD目录下会多出一些文件,其中HFNAMD就是可执行文件,如下图所示:
二、软件使用教程
我们以TiO2为例演示一下计算步骤。
1.gw计算
(1)首先利用VASP进行自洽计算,准备输入文件INCAR,POSCAR,POTCAR,KPOINTS,提交任务。具体设置如下:
INCAR:
SYSTEM = /home/wal/tio2_CO2 Startparameter for this Run: NWRITE = 2 ISTART = 0 ICHARG = 2 LCHARG = .TRUE. LWAVE = .TRUE. NCORE = 4 KPAR = 2 Electronic Relaxation PREC = High ENCUT = 400 ISPIN = 1 NBANDS = 210 NELMIN = 5 EDIFF = 1E-08 LREAL = .FALSE. IALGO = 38 GGA = PE Ionic Relaxation EDIFFG = -5E-03 IBRION = -1 ISIF = 0 NSW = 0 POTIM = 0.2 DOS Related values ISMEAR = 0 SIGMA = 0.05 LORBIT = 11 LOPTICS = .TRUE.
POSCAR:
O2Ti1
1.0
4.6723895073 0.0000000000 0.0000000000
0.0000000000 4.6723895073 0.0000000000
0.0000000000 0.0000000000 3.0231277943
Ti O
2 4
Direct
0.000000000 0.000000000 0.000000000
0.500000000 0.500000000 0.500000000
0.305055708 0.305055708 0.000000000
0.694944322 0.694944322 0.000000000
0.194944292 0.805055678 0.500000000
0.805055678 0.194944292 0.500000000
POTCAR和KPOINTS这里省略。
(2)同时还需要计算一下能带,大家应该都会,这里省略。
(3)g0w0计算,INCAR设置如下:
SYSTEM = /home/wal/tio2_CO2 Startparameter for this Run: NWRITE = 2 ISTART = 1 LCHARG = .TRUE. LWAVE = .TRUE. KPAR = 4 Electronic Relaxation PREC = High ENCUT = 400 ISPIN = 1 NBANDS = 210 EDIFF = 1E-06 LREAL = .FALSE. GGA = PE Ionic Relaxation EDIFFG = -5E-03 IBRION = -1 ISIF = 0 NSW = 0 POTIM = 0.2 DOS Related values ISMEAR = 0 SIGMA = 0.05 LORBIT = 11 GW LOPTICS = .TRUE. PRECFOCK = N NELM = 1 ALGO = GW0 NOMEGA = 50
2.计算分子动力学
在计算分子动力学之前需要先进行扩胞,将需要研究的能带折叠到Gamma点,然后结构优化删除速度信息,我们现在计算的这个体系刚好在gamma点,就不需要这一步。
(1)分子动力学NVT
准备INCAR如下:
SYSTEM = /home/wal/tio2_CO2 Startparameter for this Run: NWRITE = 2 ISTART = 0 ICHARG = 1 LCHARG = .TRUE. LWAVE = .TRUE. NCORE = 8 # KPAR = 2 Electronic Relaxation PREC = High ENCUT = 400 ISPIN = 1 # NBANDS = 240 NELMIN = 5 EDIFF = 1E-06 LREAL = .FALSE. IALGO = 38 GGA = PE Ionic Relaxation EDIFFG = -5E-03 IBRION = 0 SMASS = -1 ISYM = 0 NSW = 1000 POTIM = 1 NBLOCK = 4 TEBEG=300 TEEND=300 DOS Related values ISMEAR = 0 SIGMA = 0.05 LORBIT = 11
计算完之后可以画出温度随时间的变化图,可以使用以下脚本,来自郑奇靖老师的网站:
staff.ustc.edu.cn/~zqj/assets/hefei-namd/scripts/vaspT
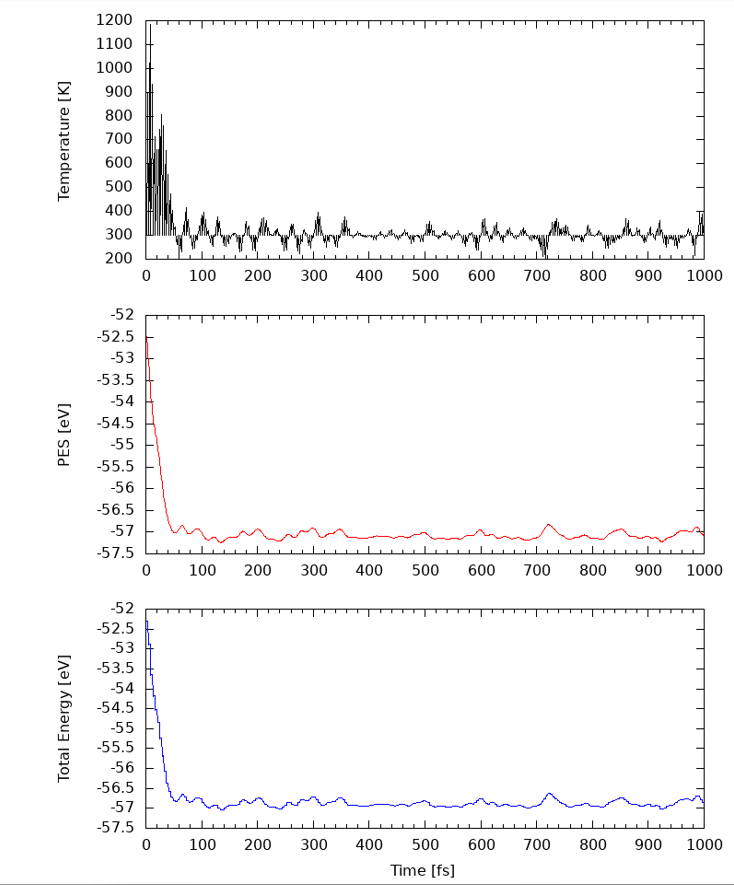
当温度平衡时,可以进行下一步
(2)分子动力学NVE
将上一步的CONTCAR复制到新的文件夹,我们命名为nve,并改为POSCAR
修改INCAR:SMASS=-3,NBLOCK=1,删掉TEBEG、TEEND:
SYSTEM = /home/wal/tio2_CO2 Startparameter for this Run: NWRITE = 2 ISTART = 0 ICHARG = 1 LCHARG = .TRUE. LWAVE = .TRUE. NCORE = 8 # KPAR = 2 Electronic Relaxation PREC = High ENCUT = 400 ISPIN = 1 # NBANDS = 240 NELMIN = 5 EDIFF = 1E-06 LREAL = .FALSE. IALGO = 38 GGA = PE Ionic Relaxation EDIFFG = -5E-03 IBRION = 0 SMASS = -3 ISYM = 0 NSW = 1000 POTIM = 1 NBLOCK = 1 DOS Related values ISMEAR = 0 SIGMA = 0.05 LORBIT = 11
nve计算结束之后,准备python脚本xdat2pos.py,脚本内容如下:
import numpy as np
import os
##############################################################################
def xdat2pos(xdatfile='XDATCAR', dir='run/'):
headfile = open(xdatfile, 'r')
headlines = headfile.readlines()[:7]
headfile.close()
head = ''.join(headlines) + 'Direct'
print(head)
# total = np.array(headlines[6].split())
# total.dtype = 'float'
# total = total.sum()
total = np.array(headlines[6].split(), dtype=int).sum()
#print(total)
xdat = np.loadtxt(xdatfile, skiprows=7, comments='D')
N = xdat.shape[0] / total
#print(N)
beginNO = 900 #not include
endNO = N
os.mkdir(dir)
for i in np.arange(beginNO, endNO) + 1:
ii = '%04d' % (i - beginNO)
posdir = dir + ii
os.mkdir(posdir)
posfile = posdir + '/POSCAR'
print(posfile)
np.savetxt(posfile, xdat[(int(i) - 1) * total:int(i) * total], fmt='%.8f',
delimiter = ' ', newline='\n ', header=head, comments='')
file = open(posfile, 'r')
midtmp = file.readlines()[:-1]
file.close()
file = open(posfile, 'w')
file.writelines(midtmp)
file.close()
############################################################################
xdat2pos()
将该脚本和nev生成的XDATCAR放在一个文件夹中,运行脚本,会产生一个run文件夹。 准备自洽计算的输入文件,incar设置如下:
SYSTEM = /home/wal/tio2_CO2 Startparameter for this Run: NWRITE = 2 ISTART = 1 ICHARG = 1 LCHARG = .TRUE. LWAVE = .TRUE. NCORE = 4 KPAR = 2 Electronic Relaxation PREC = High ENCUT = 400 ISPIN = 1 NBANDS = 42 NELMIN = 5 EDIFF = 1E-06 LREAL = .FALSE. IALGO = 38 GGA = PE Ionic Relaxation EDIFFG = -5E-03 IBRION = -1 ISIF = 0 ISYM = -1 NSW = 0 POTIM = 0.2 DOS Related values ISMEAR = 0 SIGMA = 0.05 LORBIT = 11
并用一个简单的shell脚本链接到run文件夹里面的自洽文件夹,脚本内容如下:
#!/bin/bash
for i in {0001..0100};do
cd run/$i
ln -sf ../../INCAR
ln -sf ../../KPOINTS
ln -sf ../../POTCAR
ln -sf ../../vasp.sh
sbatch < vasp.sh
cd ../..
done
目前,文件夹下有这些文件:
直接运行上面的shell脚本就开始进行自洽计算了。
自洽计算结束之后,准备脚本tdksen.py:
#!/usr/bin/env python
############################################################
import os, re
import numpy as np
from glob import glob
import matplotlib as mpl
mpl.use('agg')
mpl.rcParams['axes.unicode_minus'] = False
import matplotlib.pyplot as plt
from matplotlib.collections import LineCollection
from mpl_toolkits.axes_grid1 import make_axes_locatable
############################################################
def WeightFromPro(infile='PROCAR', whichAtom=None, spd=None):
"""
Contribution of selected atoms to the each KS orbital
"""
print(infile)
assert os.path.isfile(infile), '%s cannot be found!' % infile
FileContents = [line for line in open(infile) if line.strip()]
# when the band number is too large, there will be no space between ";" and
# the actual band number. A bug found by Homlee Guo.
# Here, #kpts, #bands and #ions are all integers
nkpts, nbands, nions = [int(xx) for xx in re.sub('[^0-9]', ' ', FileContents[1]).split()]
if spd:
Weights = np.asarray([line.split()[1:-1] for line in FileContents
if not re.search('[a-zA-Z]', line)], dtype=float)
Weights = np.sum(Weights[:,spd], axis=1)
else:
Weights = np.asarray([line.split()[-1] for line in FileContents
if not re.search('[a-zA-Z]', line)], dtype=float)
nspin = Weights.shape[0] // (nkpts * nbands * nions)
Weights.resize(nspin, nkpts, nbands, nions)
Energies = np.asarray([line.split()[-4] for line in FileContents
if 'occ.' in line], dtype=float)
Energies.resize(nspin, nkpts, nbands)
if whichAtom is None:
return Energies, np.sum(Weights, axis=-1)
else:
# whichAtom = [xx - 1 for xx in whichAtom]
return Energies, np.sum(Weights[:,:,:,whichAtom], axis=-1)
def parallel_wht(runDirs, whichAtoms, nproc=None):
'''
calculate localization of some designated in parallel.
'''
import multiprocessing
nproc = multiprocessing.cpu_count() if nproc is None else nproc
pool = multiprocessing.Pool(processes=nproc)
results = []
for rd in runDirs:
res = pool.apply_async(WeightFromPro, (rd + '/PROCAR', whichAtoms, None,))
results.append(res)
enr = []
wht = []
for ii in range(len(results)):
tmp_enr, tmp_wht = results[ii].get()
enr.append(tmp_enr)
wht.append(tmp_wht)
return np.array(enr), np.array(wht)
############################################################
# calculate spatial localization
############################################################
nsw = 100
dt = 1.0
#gapdiff = 1.70750
gapdiff = 0
nproc = 8
prefix = './run/'
runDirs = [prefix + '/{:04d}'.format(ii + 1) for ii in range(nsw)]
# which spin, index starting from 0
whichS = 0
# which k-point, index starting from 0
whichK = 0
# which atoms, index starting from 0
whichA = np.arange(2)
# whichB = range(54)
Alabel = r'Ti'
Blabel = r'O'
if os.path.isfile('all_wht.npy'):
Wht = np.load('all_wht.npy')
Enr = np.load('all_en.npy')
else:
# for gamma point version, no-spin
Enr, Wht = parallel_wht(runDirs, whichA, nproc=nproc)
Enr = Enr[:, whichS,whichK, :]
Wht = Wht[:, whichS,whichK, :]
# Enr, Wht1 = parallel_wht(runDirs, whichA, nproc=nproc)
# Enr, Wht2 = parallel_wht(runDirs, whichB, nproc=nproc)
# Enr = Enr[:, whichS,whichK, :]
# Wht1 = Wht1[:, whichS,whichK, :]
# Wht2 = Wht2[:, whichS,whichK, :]
# Wht = Wht1 / (Wht1 + Wht2)
np.save('all_wht.npy', Wht)
np.save('all_en.npy', Enr)
############################################################
fig = plt.figure()
fig.set_size_inches(4.8, 3.6)
########################################
ax = plt.subplot()
nband = Enr.shape[1]
T, dump = np.mgrid[0:nsw:dt, 0:nband]
sFac = 8
Enr[Enr > 6] += gapdiff #gw-gapdiff
############################################################
# METHOD 1.
############################################################
# use scatter to plot the band
# ax.scatter( T, Enr, s=Wht / Wht.max() * sFac, color='red', lw=0.0, zorder=1)
# for ib in range(nband):
# ax.plot(T[:,ib], Enr[:,ib], lw=0.5, color='k', alpha=0.5)
############################################################
# METHOD 2.
############################################################
# use colored scatter to plot the band
img = ax.scatter(T, Enr, s=1.0, c=Wht, lw=0.0, zorder=1,
vmin=Wht.min(),
vmax=Wht.max(),
cmap='seismic')
# for ib in range(nband):
# ax.plot(T[:,ib], Enr[:,ib], lw=0.5, color='k', alpha=0.5)
divider = make_axes_locatable(ax)
ax_cbar = divider.append_axes('right', size='5%', pad=0.02)
cbar = plt.colorbar(img, cax=ax_cbar,
orientation='vertical')
cbar.set_ticks([Wht.min(), Wht.max()])
cbar.set_ticklabels([Blabel, Alabel])
############################################################
# METHOD 3.
############################################################
# # use color strip to plot the band
#
# LW = 1.0
# DELTA = 0.3
# norm = mpl.colors.Normalize(vmin=Wht.min(),
# vmax=Wht.max())
# # create a ScalarMappable and initialize a data structure
# s_m = mpl.cm.ScalarMappable(cmap='jet_r', norm=norm)
# s_m.set_array([Wht])
#
# x = np.arange(0, nsw, dt)
# # for iband in range(nband):
# for iband in range(100, 110):
# print('Processing band: {:4d}...'.format(iband))
# y = Enr[:,iband]
# z = Wht[:,iband]
#
# ax.plot(x, y,
# lw=LW + 2 * DELTA,
# color='gray', zorder=1)
#
# points = np.array([x, y]).T.reshape(-1, 1, 2)
# segments = np.concatenate([points[:-1], points[1:]], axis=1)
# lc = LineCollection(segments,
# colors=[s_m.to_rgba(ww) for ww in (z[1:] + z[:-1])/2.]
# )
# # lc.set_array((z[1:] + z[:-1]) / 2)
# lc.set_linewidth(LW)
# ax.add_collection(lc)
#
# divider = make_axes_locatable(ax)
# ax_cbar = divider.append_axes('right', size='5%', pad=0.02)
# cbar = plt.colorbar(s_m, cax=ax_cbar,
# # ticks=[Wht.min(), Wht.max()],
# orientation='vertical')
# cbar.set_ticks([Wht.min(), Wht.max()])
# cbar.set_ticklabels([Alabel, Blabel])
ax.set_xlim(0, nsw)
ax.set_ylim(3.5, 8.5+gapdiff)
ax.set_xlabel('Time [fs]', fontsize=None, labelpad=5)
ax.set_ylabel('Energy [eV]', fontsize=None, labelpad=8)
ax.tick_params(which='both', labelsize='x-small')
########################################
plt.tight_layout(pad=0.2)
plt.savefig('dft.png', dpi=360)
#plt.savefig('gw.png', dpi=360)
目前,文件夹下有以下文件:
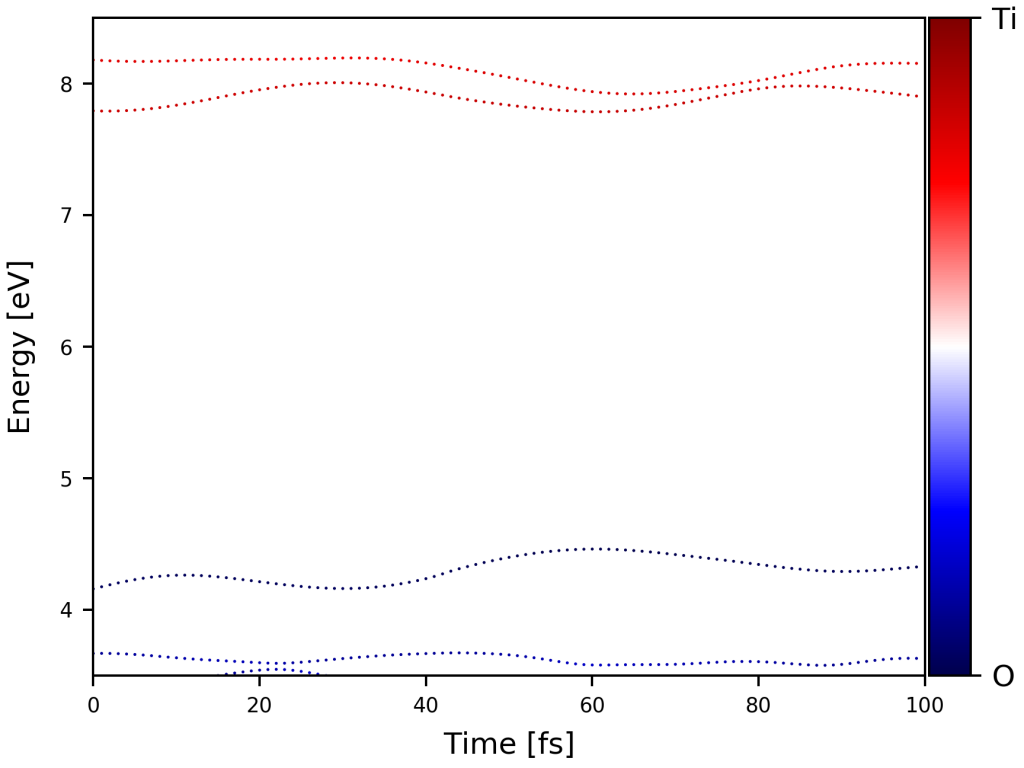
直接运行脚本,得到下图:
3.计算NAMD
前面都是namd计算的准备工作,最后一步进行namd的计算。
首先准备输入文件:input和inicon
input:
// basic
taskmod = fssh // task mode choose, now supported: fssh, dish, dcsh, bse, spinor
carrier = exciton // supported: electron(default) hole exciton
dftcode = vasp // only supported is vasp currently
vaspver = 4 // VASP version 2-5.2.x, 4-5.4.x, 6-6.x
vaspbin = std // supported: gam std ncl
//pawpsae = ae // for paw choose, ps("pseudo") or ae("all-electron")
//sexpikr = 0 // 0-calc Ekrij in momory, 1-store Ekrij in disk
ispinor = 0 // 0-Bloch 1-spinor
memrank = 1 // 1-high(default), 2-medium, >=3-low
//probind = 1 // # of processes(usually nodes) binded. Default covers one node, if memrank >= 3, probind = ALL processes(nodes)
runhome = ../md/run/ // all scf directories are listed in runhome
ndigits = 4 // # of digits for scf directories, e.g. 00136 if ndigits = 5
totstru = 100 // if totstru = 0, code will combine previous calculations
//strubeg = 1 // if set the two tags
//struend = 10 // totstru forced equals to struend - strubeg + 1
// basis sets space
numspns = 1 // numspns = 1 or 2
allspns = 0 // 0 or 1 if numspns = 1; 0 0, 0 1, 1 1 if numspns = 2
numkpts = 0 // 0 for ALL
kpoints = 3 5 6 9
bandtop = 20 20
bandmax = 18 18
bandmin = 11 11
bandbot = 8 8
condmax = 28
condmin = 25
valemax = 24
valemin = 22
exclude = 0 // format: num b_11 b_12 ... b_1num b_21 b_22 ... b_2num. total 1 + numspns * num
// dynamics
//hopmech = nac // the hopping mechanism, now supported: nac, nacsoc, nacbse
//intalgo = Magnus // integral algorithm, default: Magnus, test: Euler
dyntemp = 250. // the temperature of dynamics
iontime = 1.0 // ionic step time, POTIM in vasp
neleint = 1000 // number of electronic intergral, interval time = iontime / neleint
nsample = 2 // number of samples for dynamics
ntrajec = 10000 // number of trajectories for one sample
namdtim = 10 // namd time, should at least 2
nvinidc = 1 // non-vanishing initial dynamic coefficients
// exciton
epsilon = -1.0 // if < 0, use gw calculation, else screen Coulomb potential = 1/epsilon * 1/r
wpotdir = ../gw/g0w0/
iswfull = 0 // VASP: wxxxx.tmp or wfullxxxx.tmp
//encutgw = xxx // set encutgw ONLY if there is ENCUTGW tag in vasp INCAR file
nkptsgw = 2 2 3
nkptssc = 2 2 3
gapdiff = 1.6867 // > 0: manually set; < 0: automatically by wpotdir/OUTCAR
//dynchan = 1.0 1.0 1.0 1.0 // dynamics channel scales, size of 4, for K^d, K^x, eph, radiation
lrecomb = 1 // recombination option: 0-not recombine, 1-nonradiative, 2-radiative, 3-both
//bsedone = 0 // = 1 if bse was calculated before and
// make sure tmpDirect/ and tmpExchange/ exist in calculation directory
// addtional info
// "spinor2" > 0 means some spinor plane-wave will be rebuilt
//spinor2 = N // the following "N" lines specify rebuilt kpoints
//kpt1 kpt2 kpt3 kpt4 // example for spinor2 = 4, kpt_i = 1, 2, 3, ...
// usually kpt1 = 1 with N = 1 for Gamma is enough
inicon:
17 0 1 25 24 42 0 1 25 24
准备好namd的提交脚本,直接运行namd,我的提交脚本内容如下:
#!/bin/bash #SBATCH -J name #SBATCH -p bcores48 #SBATCH -n 48 #SBATCH --reservation=qc119_30 module load gcc/7.5.0 source /data/software/intel/2022u2/setvars.sh intel64 ulimit -s unlimited mpirun -np 48 /data/home/qc119/sourcecode/paraHFNAMD/HFNAMD
此处需要特别注意,加载的intel编译器需要和你安装namd时的编译器匹配,并且加载的gcc也要匹配。
No Comments
Leave a comment Cancel