








我们以CrI3这个结构为例,来进行基组优化
首先准备三个文件,分别命名为scf.input,systemfile_Cr.txt,basefile_I.txt,后面两个分别放在相同命名的文件夹内
calculation.name = basisOptimization %计算类型
calculation.basisOptimization.basisInputFiles = {'./basefile_Cr/basefile_Cr.txt','./basefile_I/basefile_I.txt'}
calculation.basisOptimization.basisFileNames = {'./Cr_DZP_PBE.nad','./I_DZP_PBE.nad'}
calculation.basisOptimization.systemInputFiles= {'./systemfile/systemfile.txt'}%系统文件位置
calculation.basisOptimization.simplex.MaxFunEvals = 1000
calculation.basisOptimization.simplex.TolFun = 1e-3
calculation.basisOptimization.constrainType = hard
calculation.basisOptimization.history = true
在systemfile目录下,systemfile.txt文件设置如下:
system.name = 'CrI3'
system.spinType = 'NoSpin' %自旋类型
system.atomCoordinateFormat = 'fractional'
system.centralCellVectors =...
[ [7.09082 0 0]', ...
[-3.54541 6.14083 0]', ...
[0 0 37.975]' ]
system.atomBlock = 8 %原子个数
AtomType X Y Z OrbitalType
Cr 0.3183960020000001 0.6591969730000002 0.4951409999999967 DZP_PBE
Cr 0.6517289880000021 0.3258639869999982 0.4951480029999971 DZP_PBE
I 0.3448700009999968 0.3522259889999972 0.4532969890000018 DZP_PBE
I 0.6253659730000010 0.9926429989999974 0.4532969890000018 DZP_PBE
I 0.9849500059999983 0.6327229740000035 0.4532969890000018 DZP_PBE
I 0.9849939939999999 0.3521119950000013 0.5369809869999997 DZP_PBE
I 0.6254799960000028 0.6328819989999985 0.5369809869999997 DZP_PBE
I 0.3447110059999972 0.9925990099999993 0.5369809869999997 DZP_PBE
End
calculation.name = {'SCF','BS'} %计算内容
calculation.control.logOutputLevel = [0 2]
calculation.control.lengthUnit = 'Ang'
calculation.control.energyUnit = 'eV'
calculation.realspacegrids.E_cutoff = 3000 eV
calculation.k_spacegrids.L_cutoff = 25 Ang
calculation.xcFunctional.Type = 'GGA_PBE96' %交换关联泛函
calculation.occupationFunction.temperature = 300
calculation.SCF.maximumSteps = 100
calculation.SCF.startingMode = 'H'
calculation.SCF.mixingMode = 'H'
calculation.SCF.mixMethod = Pulay
calculation.SCF.mixRate = 0.01
calculation.SCF.monitoredVariableName = {'hMatrix', 'rhoMatrix', 'totalEnergy', 'gridCharge','orbitalCharge'}
calculation.SCF.convergenceCriteria = {1e-5, 1e-5, 1e-5, [],[]}
calculation.bandStructure.primitiveCellVectors =...
[ [7.09082 0 0]', ...
[-3.54541 6.14083 0]', ...
[0 0 37.975]' ]
calculation.bandStructure.symmetryKPoints = {'M', 'G', 'K', 'M'} %高对称点
calculation.bandStructure.numberOfKPoints = [60,36,48] %高对称点取点间隔
calculation.bandStructure.coordinatesOfTheSymmetryKPoints = [-0.50000 0.50000 0.00000; 0.00000 0.00000 0.00000; -0.33333 0.66667 0.00000; -0.50000 0.50000 0.00000]' %高对称点坐标
calculation.bandStructure.plot = true %是否绘制能带图
如果要考虑自旋极化,则将system.atomCoordinateFormat = 'fractional' ,并且在systemfile.txt文件的原子坐标后面加上一列,如下:
system.atomCoordinateFormat = 'fractional' AtomType X Y Z SpinPolarization OrbitalType Cr 0.3183960020000001 0.6591969730000002 0.4951409999999967 1 DZP_PBE Cr 0.6517289880000021 0.3258639869999982 0.4951480029999971 1 DZP_PBE I 0.3448700009999968 0.3522259889999972 0.4532969890000018 1 DZP_PBE I 0.6253659730000010 0.9926429989999974 0.4532969890000018 1 DZP_PBE I 0.9849500059999983 0.6327229740000035 0.4532969890000018 1 DZP_PBE I 0.9849939939999999 0.3521119950000013 0.5369809869999997 1 DZP_PBE I 0.6254799960000028 0.6328819989999985 0.5369809869999997 1 DZP_PBE I 0.3447110059999972 0.9925990099999993 0.5369809869999997 1 DZP_PBE
basefile_Cr.txt设置如下
% What do you want to do? %计算内容
(1) I want to calculate all electron atom: yes
(2) I want to generate pseudopotential: yes
(3) I want to check log derivative: yes
(4) I want to generate fireball basis: yes
(5) I want to export basis data file: yes
%(1) Parameters for calculate all electron atom %解全电子参数
AllElectron.Atom = 'Cr'
AllElectron.Shell.Core = '1s2 2s2 2p6 3s2 3p6'
AllElectron.Shell.Valence = '4s1 4p0 3d5 4f0'
AllElectron.XCfunctional = 'GGA_PBE96'
%XCfunctional options: GGA_LYP88, GGA_PBE96, GGA_REVPBE98
%XCfunctional options: GGA_RPBE99, LSDA_PW92, LSDA_PZ81
%(2) Parameters for generate pseudopotential %产生赝势参数
PseudoAtom.CoreRadius = [2.25, 2.25, 1.85, 2.25]
PseudoAtom.PartialCore.Method = 'sin-scheme'
PseudoAtom.PartialCore.Radius = 0.80
%(3) Parameters for check log derivative
LogDerivative.type = 'AEvsPSvsKB'; %'AEvsPSvsKB', 'AEvsPS', 'PSvsKB'
LogDerivative.E = linspace(-3,3,1001)
LogDerivative.R = 4
LogDerivative.L = 0
%(4) Parameters for generate fireball basis %产生基组参数
NanoOrbital.BasisSetType = 'Confined'
NanoOrbital.ChargeTransfer = 0.00982372424916
NanoOrbital.ConfinementType = 'nano-1'
NanoOrbital.ConfinedOrbital{1} = {0, [6.69101209544 5.43941652691], [1.77320206206], [4.66519392626]}
NanoOrbital.ConfinedOrbital{2} = {1, [6.91094767999], [2.08665520787], [28.3879448926]}
NanoOrbital.ConfinedOrbital{3} = {2, [5.05601555594 3.73179379025], [1.62304615656], [3.99302014278]}'
BasisDataFile.format = 'nano'
basefile_I.txt设置如下
% What do you want to do? %计算内容
(1) I want to calculate all electron atom: yes
(2) I want to generate pseudopotential: yes
(3) I want to check log derivative: yes
(4) I want to generate fireball basis: yes
(5) I want to export basis data file: yes
%(1) Parameters for calculate all electron atom %解全电子参数
AllElectron.Atom = 'I'
AllElectron.Shell.Core = '1s2 2s2 2p6 3s2 3p6 3d10 4s2 4p6 4d10'
AllElectron.Shell.Valence = '5s2 5p5 5d0 4f0'
AllElectron.XCfunctional = 'GGA_PBE96'
%XCfunctional options: GGA_LYP88, GGA_PBE96, GGA_REVPBE98
%XCfunctional options: GGA_RPBE99, LSDA_PW92, LSDA_PZ81
%(2) Parameters for generate pseudopotential %产生赝势参数
PseudoAtom.CoreRadius = [1.50, 1.85, 2.05, 1.85]
PseudoAtom.PartialCore.Method = ''
PseudoAtom.PartialCore.Radius = 0.00
%(3) Parameters for check log derivative
LogDerivative.type = 'AEvsPSvsKB'; %'AEvsPSvsKB', 'AEvsPS', 'PSvsKB'
LogDerivative.E = linspace(-3,3,1001)
LogDerivative.R = 4
LogDerivative.L = 0
%(4) Parameters for generate fireball basis %产生基组参数
NanoOrbital.BasisSetType = 'PAO'
NanoOrbital.EnergyShift = 30
NanoOrbital.SplitNorm = 0.25
NanoOrbital.PseudoOrbital{1} = {0, [0 0], 0}
NanoOrbital.PseudoOrbital{2} = {1, [0 0], 1}'
BasisDataFile.format = 'nano'
提交命令: nohup matlab -nodisplay -r "nanodcal scf.input" &
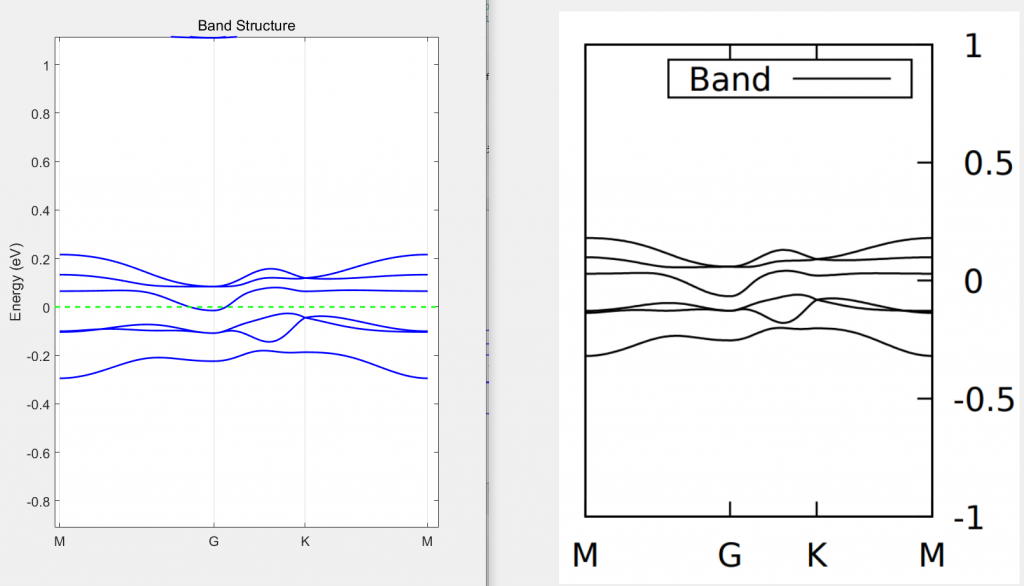
画出能带后结果如下,左图为基组优化的结果,右图为vasp计算的结果,可以看到结果匹配比较好:
官网的黑磷例子:
calculation.name = basisOptimization %计算类型
calculation.basisOptimization.basisInputFiles = {'./basefile/basefile.txt'} %基组文件位置
calculation.basisOptimization.basisFileNames = {'./P_DZP_PBE.nad'} %基组名称
calculation.basisOptimization.systemInputFiles= {'./systemfile/systemfile.txt'}%系统文件位置
calculation.basisOptimization.simplex.MaxFunEvals = 1000
calculation.basisOptimization.simplex.TolFun = 1e-3
calculation.basisOptimization.constrainType = hard
calculation.basisOptimization.history = true
system.name = 'P'
system.spinType = 'NoSpin' %自旋类型
system.atomCoordinateFormat = 'fractional'
system.centralCellVectors = [3.30,4.62,17.23]' %晶格矢量
system.atomBlock = 4 %原子个数
AtomType X Y Z OrbitalType
P 0.4999999919999993 0.0002000000000000 0.4336145293286719 DZP_PBE
P 0.0000000000000000 0.3216245004326908 0.4336145293286719 DZP_PBE
P 0.0000000000000000 0.4998040605673140 0.5558108916713280 DZP_PBE
P 0.4999999919999993 0.8216245134326883 0.5558108916713280 DZP_PBE
End
calculation.name = {'SCF','BS'} %计算内容
calculation.control.logOutputLevel = [0 2]
calculation.control.lengthUnit = 'Ang'
calculation.control.energyUnit = 'eV'
calculation.realspacegrids.E_cutoff = 3000 eV
calculation.k_spacegrids.L_cutoff = 25 Ang
calculation.xcFunctional.Type = 'GGA_PBE96' %交换关联泛函
calculation.occupationFunction.temperature = 300
calculation.SCF.maximumSteps = 100
calculation.SCF.startingMode = 'H'
calculation.SCF.mixingMode = 'H'
calculation.SCF.mixMethod = Pulay
calculation.SCF.mixRate = 0.01
calculation.SCF.monitoredVariableName = {'hMatrix', 'rhoMatrix', 'totalEnergy', 'gridCharge','orbitalCharge'}
calculation.SCF.convergenceCriteria = {1e-5, 1e-5, 1e-5, [],[]}
calculation.bandStructure.primitiveCellVectors = [3.30,4.62,17.23]' %能带计算原胞矢量
calculation.bandStructure.symmetryKPoints = {'G', 'X', 'Z', 'Y', 'G'} %高对称点
calculation.bandStructure.numberOfKPoints = [50,51,51,51] %高对称点取点间隔
calculation.bandStructure.coordinatesOfTheSymmetryKPoints = [0 0 0; 0 0.5 0; -0.5 0.5 0; 0 -0.5 0; 0 0 0]' %高对称点坐标
calculation.bandStructure.plot = true %是否绘制能带图
% What do you want to do? %计算内容
(1) I want to calculate all electron atom: yes
(2) I want to generate pseudopotential: yes
(3) I want to check log derivative: yes
(4) I want to generate fireball basis: yes
(5) I want to export basis data file: yes
%(1) Parameters for calculate all electron atom %解全电子参数
AllElectron.Atom = 'P'
AllElectron.Shell.Core = '1s2 2s2 2p6'
AllElectron.Shell.Valence = '3s2 3p3 3d0 4f0'
AllElectron.XCfunctional = 'GGA_PBE96'
%XCfunctional options: GGA_LYP88, GGA_PBE96, GGA_REVPBE98
%XCfunctional options: GGA_RPBE99, LSDA_PW92, LSDA_PZ81
%(2) Parameters for generate pseudopotential %产生赝势参数
PseudoAtom.CoreRadius = [1.50, 1.75, 1.85, 1.85]
PseudoAtom.PartialCore.Method = 'sin-scheme'
PseudoAtom.PartialCore.Radius = 1.10
%(3) Parameters for check log derivative
LogDerivative.type = 'AEvsPSvsKB'; %'AEvsPSvsKB', 'AEvsPS', 'PSvsKB'
LogDerivative.E = linspace(-3,3,1001)
LogDerivative.R = 4
LogDerivative.L = 0
%(4) Parameters for generate fireball basis %产生基组参数
NanoOrbital.BasisSetType = 'Confined'
NanoOrbital.ChargeTransfer = 0.0035431386905
NanoOrbital.ConfinementType = 'nano-1'
NanoOrbital.ConfinedOrbital{1} ={0, [7.51218749485 3.55678760271], [1.91444329546],[2.26296289044]} %{L,[Rc1,Rc2]Ri,V0}
NanoOrbital.ConfinedOrbital{2} ={1, [7.19912115594 3.92933021476], [1.58189238251],[1.5590946047]}
NanoOrbital.ConfinedOrbital{3} ={2, [7.02107224987], [0.0948900016922], [16.399172853]}
BasisDataFile.format = 'nano'
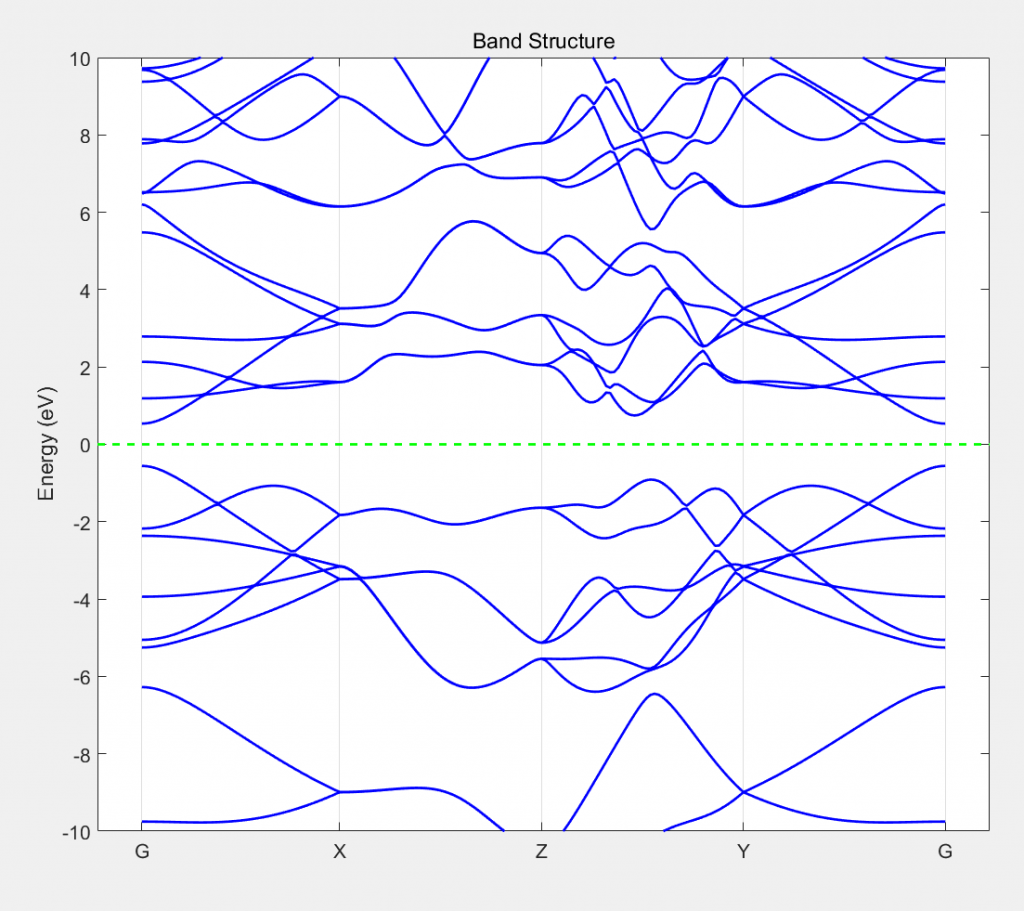
计算得到的结果用matlab画图:
No Comments
Leave a comment Cancel