class tb_model(object):
def __init__(self,dim_k,dim_r,lat=None,orb=None,per=None,nspin=1):
self._dim_k=dim_k
self._dim_r=dim_r
self._lat=np.array(lat,dtype=float)
if per==None:
self._per=list(range(self._dim_k)) #产生一个dim_k维的列表
self._nspin=nspin
self._orb=np.array(orb,dtype=float)
self._norb=self._orb.shape[0] #原子轨道的个数,shape输出几行几列,shape(0)输出行数,shape(1)输出列数
self._assume_position_operator_diagonal=True
self._nsta=self._norb*self._nspin #计算每一个k点的电子态数
if self._nspin==1: ### 产生一个列表,形如(site_energy,site_energy)
self._site_energies=np.zeros((self._norb),dtype=float)
elif self._nspin==2:
self._site_energies=np.zeros((self._norb,2,2),dtype=complex) ### 将上面的site_energy变成一个2*2的矩阵
# print(self._site_energies)
#这里相当于一个开关
self._site_energies_specified=np.zeros(self._norb,dtype=bool) #return an array filled with 0 ,and the type is bool,which means zhe value only has 0 and 1
self._site_energies_specified[:]=False #let the array is 0
self._hoppings=[]
定义函数self._val_to_block(),这个函数可以根据自旋设置哈密顿矩阵
def _val_to_block(self,val): #如果自旋为2,就从输入的参数中返回一个2*2的矩阵,如果只输入了一个实数,就假设它们是对角项
#如果输入的列表有四个元素,则假设第一个为对角项,其余三个是赛曼场方向,如果是两个数,就直接返回它
if self._nspin==1:
return val
# spinfull case
elif self._nspin==2:
# matrix to return
ret=np.zeros((2,2),dtype=complex)
#
use_val=np.array(val)
# only one number is given
if use_val.shape==():
ret[0,0]+=use_val
ret[1,1]+=use_val
# if four numbers are given
elif use_val.shape==(4,):
# diagonal
ret[0,0]+=use_val[0]
ret[1,1]+=use_val[0]
# sigma_x
ret[0,1]+=use_val[1]
ret[1,0]+=use_val[1] # sigma_y
ret[0,1]+=use_val[2]*(-1.0j)
ret[1,0]+=use_val[2]*( 1.0j)
# sigma_z
ret[0,0]+=use_val[3]
ret[1,1]+=use_val[3]*(-1.0)
# if 2 by 2 matrix is given
elif use_val.shape==(2,2):
return use_val
else:
raise Exception(\
"""\n
Wrong format of the on-site or hopping term. Must be single number, or
in the case of a spinfull model can be array of four numbers or 2x2
matrix.""")
return ret
设置在位能,即哈密顿量的对角项
def set_onsite(self,onsite_en,ind_i=None,mode="set"):
for i in range(self._norb):
self._site_energies[i]=self._val_to_block(onsite_en[i]) #fill in the array with onsite energy
self._site_energies_specified[:]=True
# print(self._site_energies)
定义跃迁项
def set_hop(self,hop_amp,ind_i,ind_j,ind_R=None,mode="set",allow_conjugate_pair=False):
hop_use=self._val_to_block(hop_amp) #由这里控制跃迁系数是矩阵还是一个数
use_index=None
new_hop=[hop_use,int(ind_i),int(ind_j),np.array(ind_R)]
if mode.lower()=="set":
# make sure we specify things only once
if use_index!=None:
raise Exception("\n\nHopping energy for this site was already specified! Use mode=\"reset\" or mode=\"add\".")
else:
self._hoppings.append(new_hop)
# print(self._hoppings)
获得哈密顿矩阵
def _gen_ham(self,k_input=None):
kpnt=np.array(k_input)
if self._nspin==1:
ham=np.zeros((self._norb,self._norb),dtype=complex)
elif self._nspin==2:
ham=np.zeros((self._norb,2,self._norb,2),dtype=complex)
# print("step one",ham)
for i in range(self._norb):
if self._nspin==1:
ham[i,i]=self._site_energies[i]
elif self._nspin==2:
ham[i,:,i,:]=self._site_energies[i]
# print("step two",ham)
for hopping in self._hoppings:
if self._nspin==1:
amp=complex(hopping[0])
elif self._nspin==2:
amp=np.array(hopping[0],dtype=complex) #如果自旋为2,跃迁系数变为一个矩阵
i=hopping[1]
j=hopping[2]
# in 0-dim case there is no phase factor
if self._dim_k>0:
ind_R=np.array(hopping[3],dtype=float)
# vector from one site to another
rv=-self._orb[i,:]+self._orb[j,:]+ind_R
# Take only components of vector which are periodic
rv=rv[self._per] #self._per是一个dim_k维的列表,例如【0,1】,这里记录了两个轨道之间的位矢,维数为dim_k维
# Calculate the hopping, see details in info/tb/tb.pdf
phase=np.exp((2.0j)*np.pi*np.dot(kpnt,rv)) #这里计算出相位,由于是分数坐标,把笛卡尔坐标基矢乘以倒格矢,归结到2pi里面
amp=amp*phase #跃迁系数乘以相位
# add this hopping into a matrix and also its conjugate
if self._nspin==1:
ham[i,j]+=amp
ham[j,i]+=amp.conjugate()
elif self._nspin==2:
ham[i,:,j,:]+=amp
ham[j,:,i,:]+=amp.T.conjugate()
# print("step three",ham)
return ham
解哈密顿量
def _sol_ham(self,ham,eig_vectors=False):
"""Solves Hamiltonian and returns eigenvectors, eigenvalues"""
# reshape matrix first
if self._nspin==1:
ham_use=ham
elif self._nspin==2:
ham_use=ham.reshape((2*self._norb,2*self._norb)) #这里将前面的列表整理成矩阵的形式
# check that matrix is hermitian
if np.max(ham_use-ham_use.T.conj())>1.0E-9:
raise Exception("\n\nHamiltonian matrix is not hermitian?!")
#solve matrix
if eig_vectors==False: # only find eigenvalues
eval=np.linalg.eigvalsh(ham_use)
# sort eigenvalues and convert to real numbers
eval=_nicefy_eig(eval)
return np.array(eval,dtype=float)
else: # find eigenvalues and eigenvectors
(eval,eig)=np.linalg.eigh(ham_use)
# transpose matrix eig since otherwise it is confusing
# now eig[i,:] is eigenvector for eval[i]-th eigenvalue
eig=eig.T
# sort evectors, eigenvalues and convert to real numbers
(eval,eig)=_nicefy_eig(eval,eig)
# reshape eigenvectors if doing a spinfull calculation
if self._nspin==2:
eig=eig.reshape((self._nsta,self._norb,2))
return (eval,eig)
将生成的高对称点代如哈密顿量里面,解哈密顿量
def solve_all(self,k_list=None,eig_vectors=False):
if not (k_list is None):
nkp=len(k_list)
ret_eval=np.zeros((self._nsta,nkp),dtype=float) #产生一个矩阵,行数为轨道数乘以自旋,列数为k点数
# print(ret_eval)
if self._nspin==1: #这里存放的是本征向量
ret_evec=np.zeros((self._nsta,nkp,self._norb),dtype=complex)
elif self._nspin==2:
ret_evec=np.zeros((self._nsta,nkp,self._norb,2),dtype=complex)
# print(ret_evec)
# ham=self._gen_ham([0.0130719 , 0.00653595])
# print(ham)
# eval=self._sol_ham(ham)
# print(eval)
for i,k in enumerate(k_list): #将k_list进行标号
# generate Hamiltonian at that point
ham=self._gen_ham(k)
# solve Hamiltonian
# print(ham)
if eig_vectors==False:
eval=self._sol_ham(ham,eig_vectors=eig_vectors)
ret_eval[:,i]=eval[:] #将解得的本征值竖着填,因为列数是k点数,行数为能带数
else:
(eval,evec)=self._sol_ham(ham,eig_vectors=eig_vectors)
ret_eval[:,i]=eval[:]
if self._nspin==1:
ret_evec[:,i,:]=evec[:,:]
elif self._nspin==2:
ret_evec[:,i,:,:]=evec[:,:,:]
# print(ret_eval)
if eig_vectors==False:
# indices of eval are [band,kpoint]
return ret_eval
else:
# indices of eval are [band,kpoint] for evec are [band,kpoint,orbital,(spin)]
return (ret_eval,ret_evec)
def readPOSCAR():
posfile = 'POSCAR'
fIn = open(posfile, 'r')
lines = fIn.readlines()
# get lat
lat = np.zeros((3, 3), dtype=np.float64)
for i in range(3):
tmp = lines[2 + i].rstrip().split() # i从0开始,加到2,将第三行到第六行的数据存放在 tmp 中
for j in range(3):
lat[i, j] = float(tmp[j])
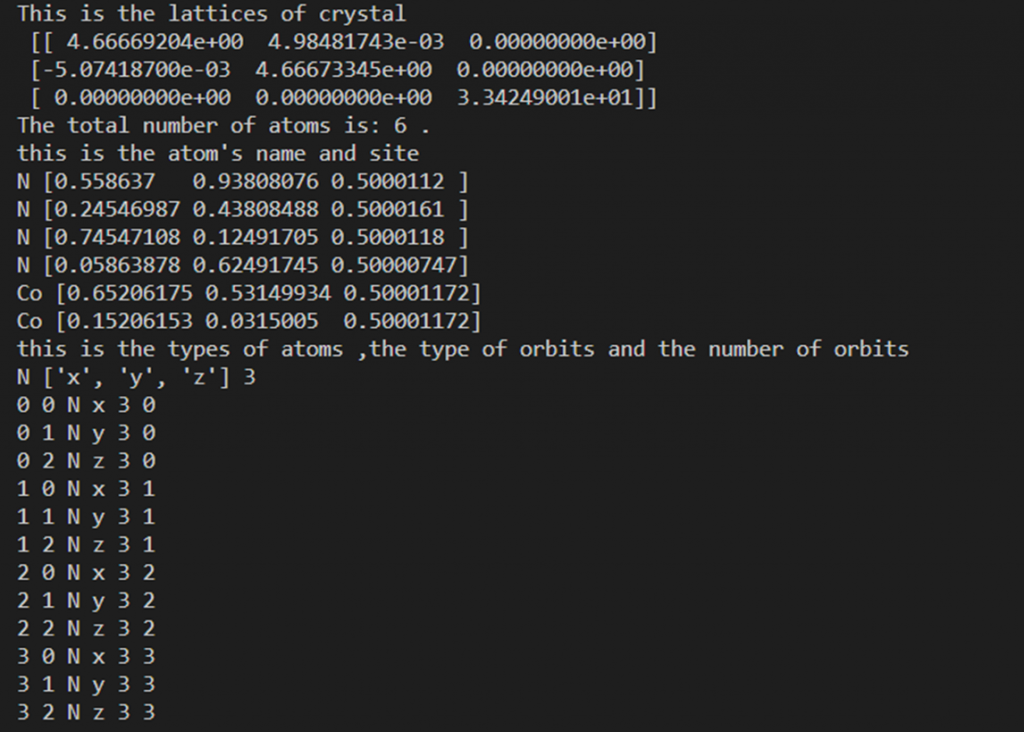
print("This is the lattices of crystal", "\n",
lat) # lat 存放基矢坐标
namelist = lines[5].rstrip().split() # 第六行为元素的名称
nTypes = len(namelist)
tmp = lines[6].rstrip().split()
# get nAtoms
numlist = [0] * nTypes
for i in range(nTypes):
numlist[i] = int(tmp[i]) # numlist 存放每种元素个数,例如【2,2,2】,natoms=6
nAtoms = sum(numlist)
print('The total number of atoms is:', nAtoms, '.')
coorlist = np.zeros((nAtoms, 3), dtype=np.float64) # 设置一个矩阵,矩阵的行数为原子的个数
for i in range(nAtoms):
tmp = lines[8 + i].rstrip().split()
for j in range(3):
coorlist[i, j] = float(tmp[j])
# print(coorlist)
# initialize arrAtom
arrAtom = list(range(nAtoms))
iCount = 0
for i in list(range(nTypes)):
for j in range(numlist[i]):
arrAtom[iCount] = Atom(namelist[i], coorlist[iCount])
iCount += 1
print("this is the atom's name and site")
for i in arrAtom:
print(i.atmType, i.coor)
orbfile = 'tb.in'
fIn = open(orbfile, 'r')
lines = fIn.readlines()
tmp = lines[1].rstrip().split()
ntypes = int(tmp[0])
typelist = list(range(ntypes))
orblist = list(range(ntypes))
norblist = list(range(ntypes))
for i in list(range(ntypes)):
tmp = lines[2 + i].rstrip().split()
ntmp = len(tmp)
norblist[i] = ntmp - 1 # norblist为轨道的个数,因为第一个是原子,所以要减1
typelist[i] = tmp[0]
orblist[i] = tmp[1:ntmp]
print("this is the types of atoms ,the type of orbits and the nember of orbits")
print(typelist[i], orblist[i], norblist[i])
nAtoms = len(arrAtom) # nAtoms为总的原子个数
# print(nAtoms)
for i in range(nAtoms):
tmp = list(arrAtom[i].atmType) # 将原子的种类存放在tmp中
# print(tmp)
# print(ntypes)
for j in range(ntypes): # 这里ntypes是需要拟合的原子种类,和上面natoms区分
# print(typelist[j])
if tmp == list(typelist[j]): # name of element matches!
ntmp = norblist[j] # 把需要拟合的轨道的个数赋值给ntmp
arrAtom[i].nOrbs = ntmp # 一共有几种轨道
# 分别为轨道的个数和轨道,注意这里也是list!!!!!
arrAtom[i].orbs = list(range(ntmp))
# print(arrAtom[i].nOrbs,arrAtom[i].orbs)
for k in list(range(ntmp)):
typetmp = list(orblist[j])
# print(typetmp[k])
arrAtom[i].orbs[k] = Orbit(
orbType=typetmp[k], iOfAtom=i, coor=arrAtom[i].coor)
# 第i个原子,第k个轨道,原子的种类,轨道的种类,一共有几个原子,在这几个中属于第几个
print(i, k, arrAtom[i].atmType, arrAtom[i].orbs[k].orbType,
arrAtom[i].nOrbs, arrAtom[i].orbs[k].iOfAtom)
# get number of orbs
nOrbits = 0
for i in range(nAtoms):
nOrbits += arrAtom[i].nOrbs # 总的轨道数
# print nOrbits
# get global coor list
arrOrb = list(range(nOrbits))
iOrb = 0
for i in range(nAtoms):
for j in range(arrAtom[i].nOrbs):
arrOrb[iOrb] = arrAtom[i].orbs[j]
arrOrb[iOrb].coor = arrAtom[i].coor # 这里定义了轨道的坐标和原子坐标相同
arrAtom[i].orbs[j].idx = iOrb
print('OO', i, j, arrAtom[i].orbs[j].idx) # 第i个原子的第j个轨道
iOrb += 1
orb = list(range(nOrbits)) # 在总的轨道数中给轨道编号,并且给出坐标,这里直接填入tb主程序
for i in range(nOrbits):
orb[i] = arrOrb[i].coor
print(orb[i])
nspin = 1
modelread = tb_model(3, 3, lat, orb, nspin=nspin)
nOrbits *= nspin # 自旋为2不需要其他设置,只需要轨道数乘以2
print(orb)
return lat, orb, nAtoms, nOrbits, arrAtom, modelread









No Comments
Leave a comment Cancel